Finasteride vs Spironolactone for Female Androgenetic Alopecia: How do side effects compare?
Comparison of Side Effects of Finasteride and Spironolactone For Women
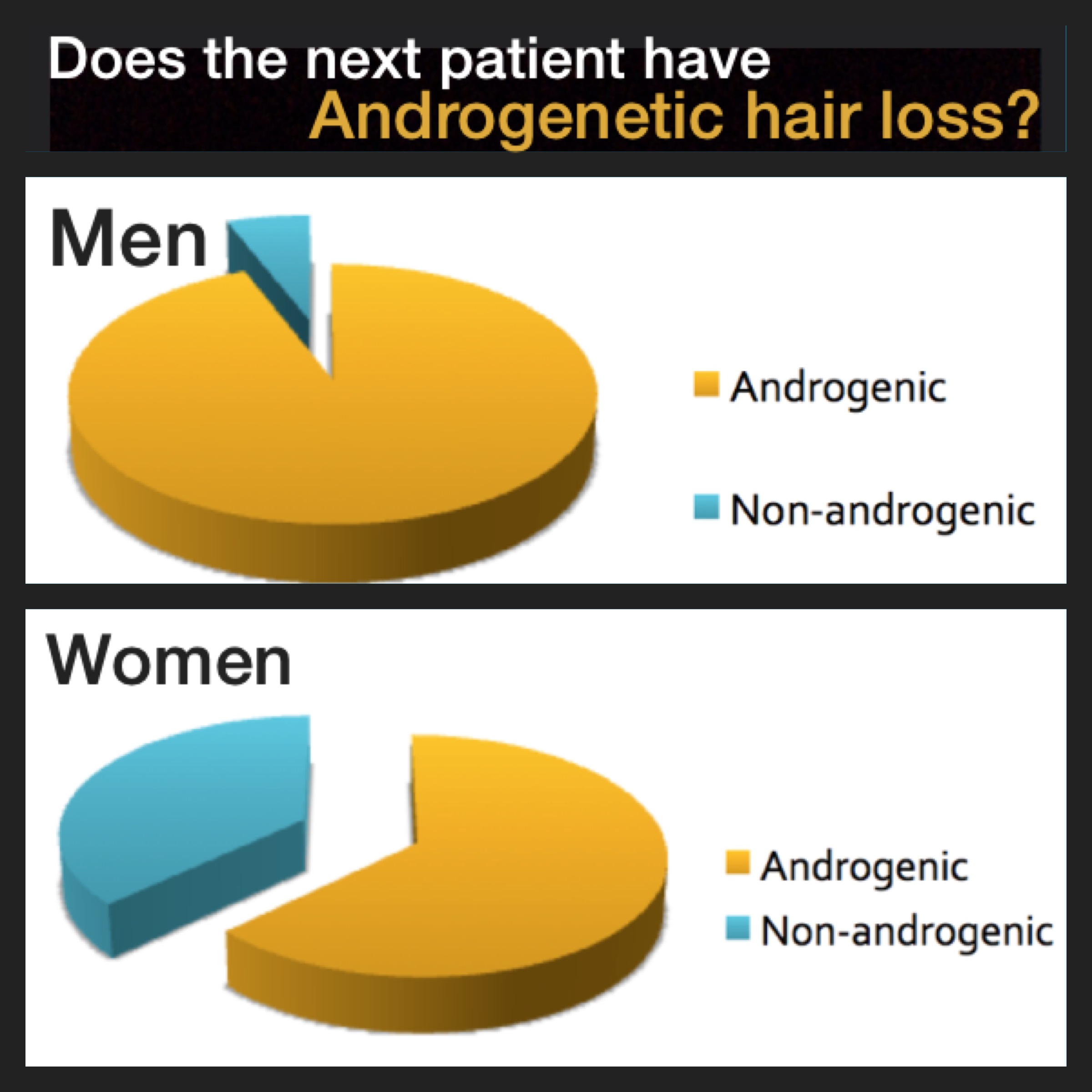
Androgenetic alopecia (AGA), also called female pattern hair loss, is common among women. By age 50, about 40 % of women will have AGA. Treatments include minoxidil, anti androgens, oral contraceptives, laser, PRP, hair transplantation, as well as others. Minoxidil remains the only formally FDA approved treatment.
There are no FDA approved anti androgens for treating female AGA although many are used off label in treating this type of hair loss. The evidence would suggest that anti-androgens are potentially the most consistently effective treatments. The antiandrogens finasteride and spironolactone are among the most commonly prescribed antiandrogens for treating female AGA. Dutasteride, bicalutamide, flutamide and cyproterone acetate are less commonly prescribed.
Finasteride vs Spironolactone: What are the side effects and how to they compare?
This article will focus on the side effects of spironolactone and finasteride. It is important for both prescribers as well as users of these medications to understand the risks and benefits of these medications before committing to use. The use of antiandrogens is lifelong when treating AGA. These medications can be helpful for treating AGA but are not appropriate for every female patent with AGA. The risks and benefits must be carefully reviewed. All anti androgens are contraindicated during pregnancy and also by any female patient who may become pregnant or is trying to conceive. Antiandrogens have the potential to cause serious harm to a developing fetus.
There have been studies of both finasteride in treating AGA and spironolactone in treating AGA. For Spironolactone, the vast majority of our understanding of side effects comes from understanding the side effects that this medication causes in women who use the medication for acne and hirsutism. There have been no good side by side comparative studies of finasteride and spironolactone in treating AGA.
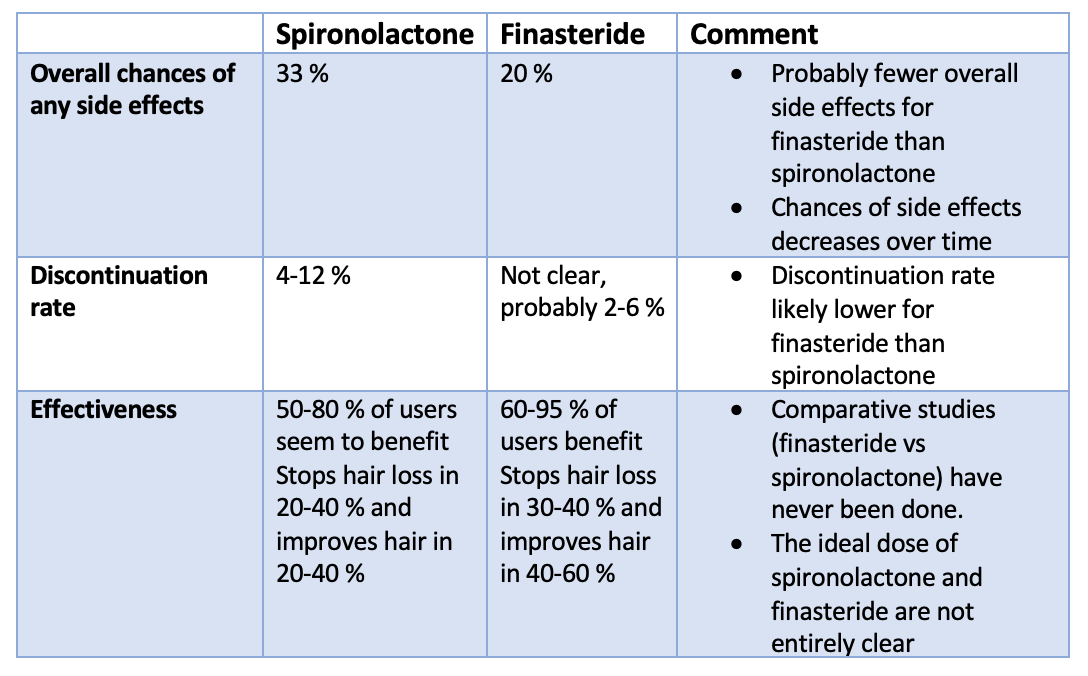
Overall, studies would suggest that finasteride probably has fewer overall side effects which contributes to its discontinuation rate being lower. However, there are many reasons that physicians may choose spironolactone over finasteride, especially in premenopausal women.
1. Overall side effect rates, Discontinuation Rates and Effectiveness
2. Non specific Effects Spironolactone vs Finasteride among Female Users
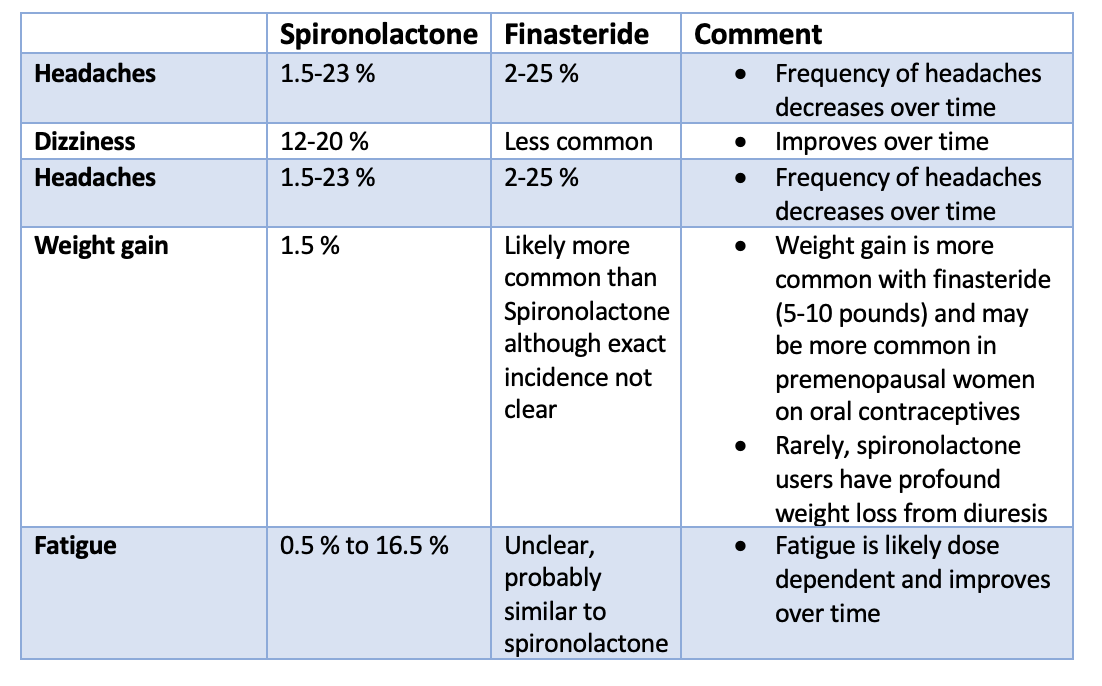
3. Specific side Effects Spironolactone vs Finasteride among Female Users
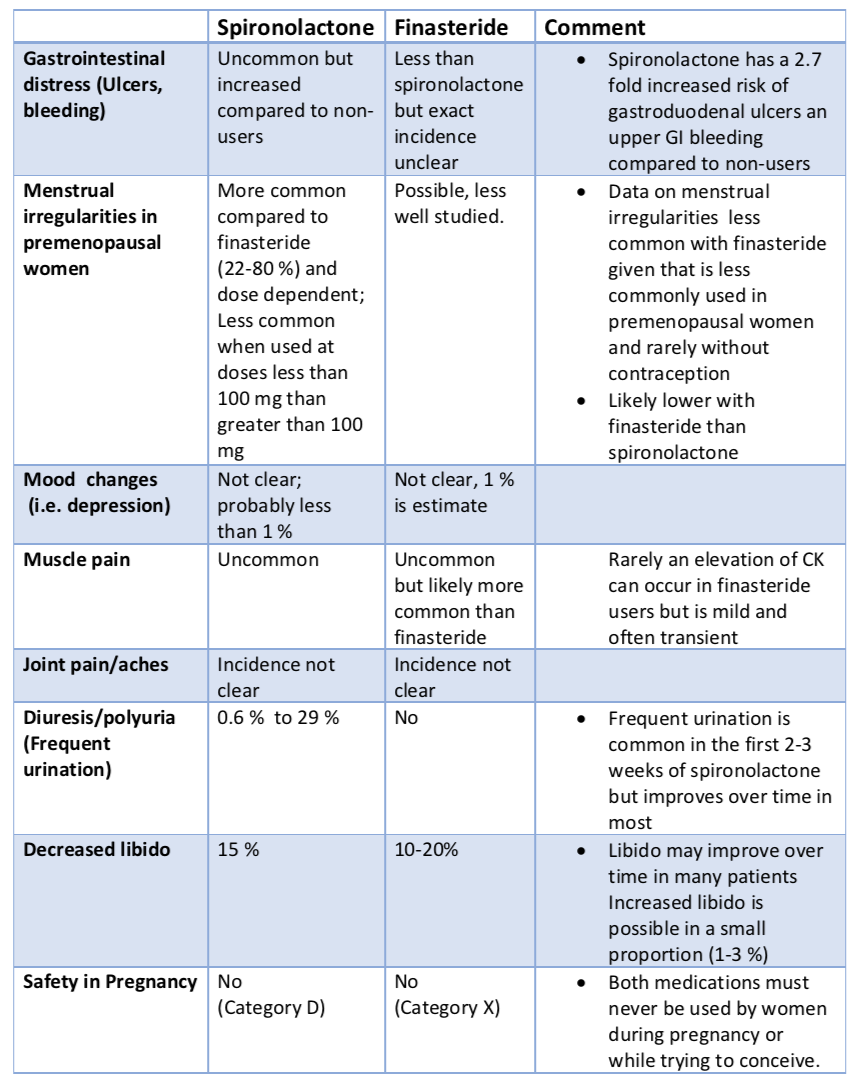
4. Side Effects Related to Breast Health

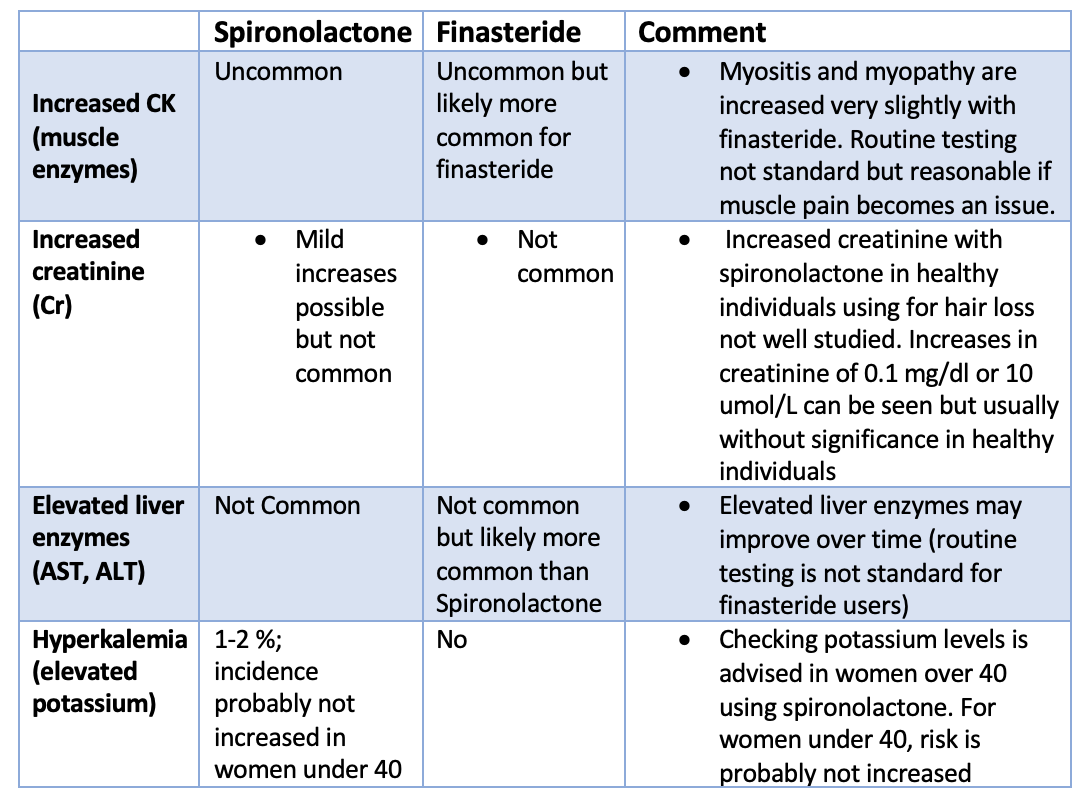
5. Laboratory Changes among Female Finasteride and Spironolactone Users.
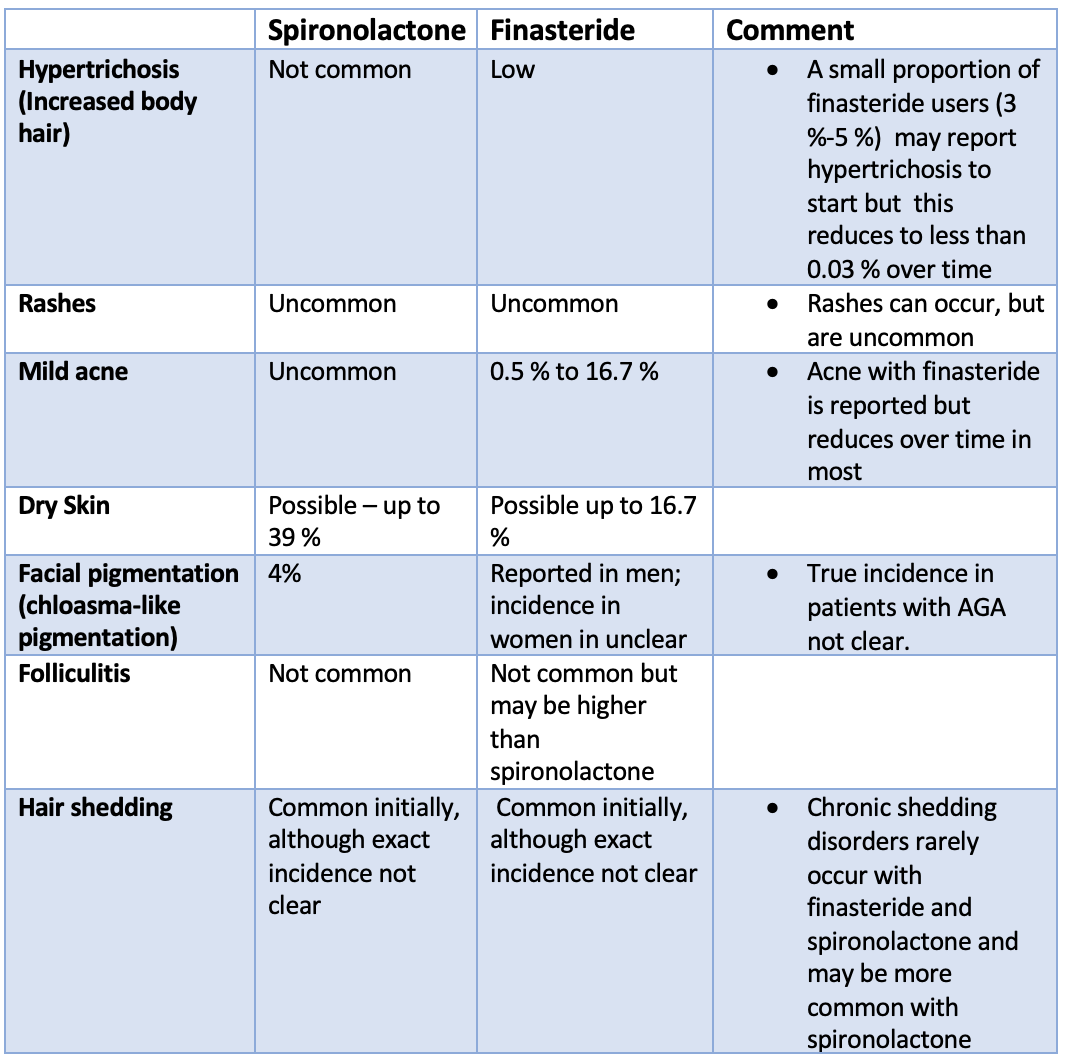
6. Dermatological Side Effects among Female Spironolactone and Finasteride Users.
Summary and Conclusion
One must understand the risks and benefits of finasteride and spironolactone if use of these medications is being considered. These medications remain options for female patients with androgenetic alopecia. A careful review of side effects with one’s physician is essential in all individuals considering these medications. There are clearly certain situations where one medication might be preferred over the other. For example, given the effects on blood pressure, the use of finasteride is often preferred in patients with issues with problematic low blood pressure.
Many of the side effects listed above improve over time. Trueb and colleagues showed showed that finateride side effects decrease over time. This incluces side effects such as libido reduction, breast tenderness, or hypertrichosis/hirsutism decrease over time. Probably, there are some adaptative hormonal changes in brain-hormonal axis or in brain perception that lead to these side effects being less and less noticed.
References
Donovan J. Spironolactone in Female AGA
Donovan J. Bicalutamide for Female AGA
Donovan J. Finasteride Use in Women: Yes or No?
Donovan J. Flutamide in Women who Don’t Respond to Spironolactone
Goodfellow A. Oral Spironolactone Improves Acne Vulgaris and Reduces Sebum Excretion.Br J Dermatol 1984 Aug;111(2):209-14.
Helfer EL et al. Side-effects of spironolactone therapy in the hirsute woman.J Clin Endocrinol Metab. 1988 Jan;66(1):208-11. doi: 10.1210/jcem-66-1-208.PMID: 3335604
Hu et al. The Efficacy and Use of Finasteride in Women: A Systematic Review. Int J Dermatol. 2019 Jul;58(7):759-776.
Hughes BR, Cunliffe W. Tolerance of spironolactone. Br J Dermatol. 1988 May;118(5):687-91. doi: 10.1111/j.1365-2133.1988.tb02571.x.PMID: 2969259
Kohler C, Tschumi K, Bodmer C, et al. Effect of finasteride 5mg (Proscar) on acne and alopecia in female patients with 72. normal serum levels of free testosterone. Gynecol Endocrinol. 2007;23:142–145.
Oliveira-Soares R, et al. 5 mg/day for Patterned Hair Loss in Premenopausal Women.Int J Trichology. 2018 Jan-Feb;10(1):48-50. doi: 10.4103/ijt.ijt_73_15.
Plovanich M et al.. Low Usefulness of Potassium Monitoring Among Healthy Young Women Taking Spironolactone for Acne. JAMA Dermatol. 2015 Sep;151(9):941-4. doi: 10.1001/jamadermatol.2015.34.
Seale LR, Eglini AN, McMichael AJ. Side Effects Related to 5 α-Reductase Inhibitor Treatment of Hair Loss in Women: A Review. J Drugs Dermatol 2016 Apr;15(4):414-9.
Shaw JC, White LE. Long-term safety of spironolactone in acne: results of an 8-year followup study. J Cutan Med Surg. 2002 Nov-Dec;6(6):541-5. doi: 10.1007/s10227-001-0152-4. Epub 2002 Sep 12.PMID: 12219252
Townsend KA, Marlowe KF. Relative safety and efficacy of finasteride for treatment of hirsutism. Ann Pharmacother. 2004 Jun;38(6):1070-3.
Trüeb RM, Swiss Trichology Study Group. Finasteride treatment of patterned hair loss in normoandrogenic postmenopausal women. Dermatology 2004;209:202-7.
Yemisci A et al. Effects and side-effects of spironolactone therapy in women with acne. J Eur Acad Dermatol Venereol. 2005 Mar;19(2):163-6. doi: 10.1111/j.1468-3083.2005.01072.x.PMID: 15752283
This article was written by Dr. Jeff Donovan, a Canadian and US board certified dermatologist specializing exclusively in hair loss.