Hormonal Changes in Late Onset Congenital Adrenal Hyperplasia
Hormonal Changed in Late Onset CAH Resemble PCOS
Late onset on “non-classic” congenital adrenal hyperplasia (LOCAH) is an uncommon genetic disorder that is most often (95 % of the time) caused by mutations in 21-hydroxylase gene. This mutation leads to reduced levels of the 21 hydroxylase enzyme. Late onset CAH from deficiencies or mutations in other genes such as 11β-hydroxylase (CYP11B1) and 3β-hydroxysteroid dehydrogenase (HSD3B2) are extremely rare.
Patients with LOCAH have CYP21A2 mutations that lead to reduced levels on the 21 hydroxylase enzyme but not a complete absence. The amount of functional 21-hydroxylase enzyme determines the severity of the disorder. Low levels of the enzyme result in low levels of hormones such as cortisol and/or aldosterone and high levels of androgens (male hormones such as testosterone and androstenedione).
As a result of low cortisol, patients may experience changes in energy levels, blood pressure, blood sugar levels, as well as impaired ability of the body to respond to stress, illness, and injury. Aldosterone plays a key role in helping the body maintain the proper level of sodium and water and helps maintain blood pressure.
Diagnosis of LOCAH
The patient's signs and symptoms may point to a possible diagnosis. Even though LOCAH has several endocrine issues (high androgens and low cortisol), generally speaking, the clinical features of LOCAH are due to the excess of the androgens. Taking a careful medical history from the patient may reveal premature pubarche (i.e. the development of pubic hair, axillary hair, and/or increased apocrine odor prior to age 8 years in girls and age 9 years in boys). Affected children may be tall and have accelerated linear growth velocity, and advanced skeletal maturation.
Although LOCAH is estimated to occur in less than 1:1000 individuals, overall about 2-9 % of all women with hyperandrogenism may have late onset CAH. It has been estimate that 5 % of all women with hirsutism have LOCAH. Other symptoms can closely resemble the constellation of symptoms seen in women with polycystic ovarian syndrome. For example, women with late onset CAH may develop a variety of symptoms including frontal baldness, hirsutism, acne, and irregular periods. Other symptoms include a delay in the timing of the very first period, early onset of pubic hair, accelerated growth, reduced final height and infertility.
In a 2000 study by Moran and colleagues, the three most common symptoms among adolescent and adult women with LOCAH were hirsutism (59%), oligomenorrhea (54%), and acne (33%). Studies in 2009 by Bidet and colleagues suggested that the initial presenting symptoms in 161 women with late onset CAH were hirsutism (78%), menstrual dysfunction (54.7%), and decreased fertility (12%).
Testing for LOCAH
Blood tests. Generally, additional testing is ordered to help confirm the diagnosis. These tests may include a blood test to measure the concentration of 17-hydroxyprogesterone (17-OHP) on day 3-5 of the menstrual cycle. Levels of 170–300 ng/dL have been found to be an excellent screening tool. These should be obtained in the morning and during the follicular (preovulatory) phase of the menstrual cycle.
The clinical features of late onset CAH in postpubertal adults may be difficult to differentiate from those of the polycystic ovary syndrome (PCOS). The following table shows some of the key differences in women with PCOS vs LOCAH. Note that even 17 OHP concentrations may be within the normal range for individuals with late onset CAH. LH levels can be elevated in LOCAH and the LH/FSH ratio above 3:1 can also sometimes be observed. Prolactin levels are sometimes modestly elevated and DHEAS, testosterone and androstenedione levels may or may not be elevated. Aldosterone and cortisol levels are often normal may be lower range . Low sodium or high potassium levels may be present in the blood and abnormalities in glucose levels might be present.
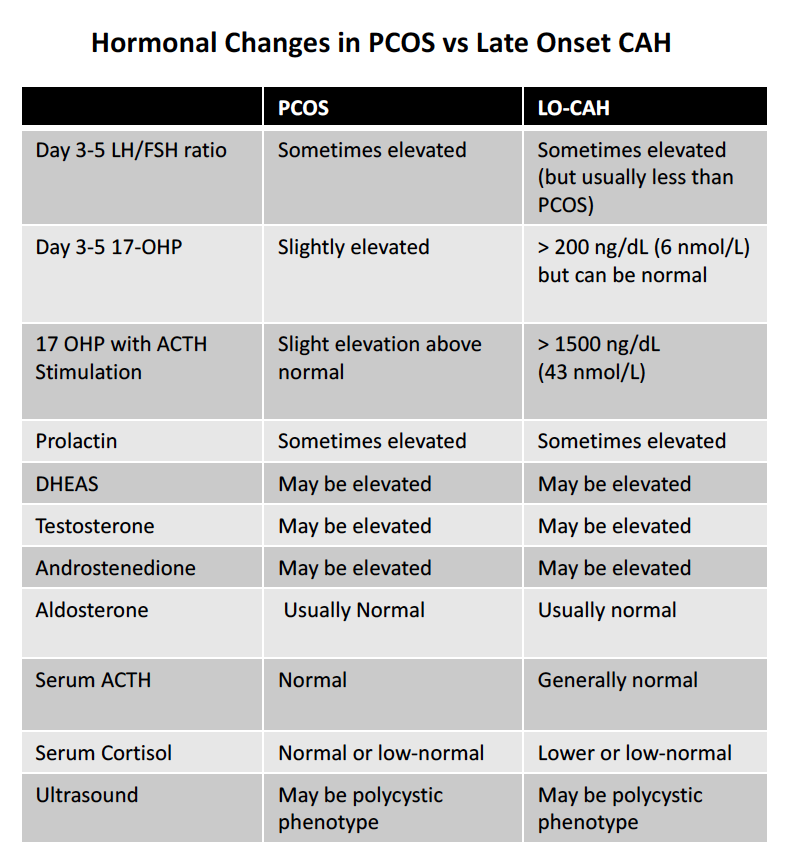
Comparison of Testing in PCOS vs Late Onset CAH. DOWNLOAD PDF VERSION
ACTH Stimulation Test. An adrenocorticotropic hormone (ACTH) stimulation test may also be ordered which involves measuring the concentration of 17-OHP in the blood before ACTH is administered to the patient and then again 60 min after ACTH is given. This test is typically conducted through an endocrinologist. The acute ACTH stimulation test remains the gold standard to confirm decreased 21-hydroxylase activity.
To perform the ACTH stimulation test, a blood sample is first collected to measure baseline hormone concentrations. Then, synthetic ACTH (Cortrosyn, 0.25 mg) is administered. A second blood sample is collected 30–60 minutes later. When the ACTH-stimulated 17-OHP value exceeds 1500 ng/dL a mutation is likely.
Ultrasound of Ovaries. Ultrasounds of the ovaries may not necessarily help differentiate PCOS from LOCAH because ultrasounds in LOCAH may show an ovarian morphology similar to polycystic ovary syndrome (PCOS) in about 50 % of patients.
Other tests
In addition to performing blood tests for day 3-5 17 OHP, other tests may be recommended by the physician caring for the patient. They include cortisol, androstenedione, testosterone, free testosterone, DHEAS, progesterone, sodium, potassium, creatinine, glucose, hemoglobin A1C. LH and FSH may also be measured. Aldosterone may be tested. Blood pressure measurements will also be obtained.
REFERENCES
56. Azziz R, Dewailly D, Owerbach D. Nonclassic adrenal hyperplasia: current concepts. Journal of Clinical Endocrinology and Metabolism. 1994;78(4):810–815.
Witchel et al. Nonclassic Congenital Adrenal Hyperplasia Int J Pediatr Endocrinol. 2010; 2010: 625105.
Moran C, Azziz R, Carmina E, et al. 21-hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. American Journal of Obstetrics and Gynecology. 2000;183(6):1468–1474.
Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. Journal of Clinical Endocrinology and Metabolism. 2009;94(5):1570–1578.
Scaroni C et al. HLA and hormonal studies in 5 patients with late-onset 21-hydroxylase deficiency syndrome (21 OHDS). Journal of Endocrinological Investigation. February 1986, Volume 9, Issue 1, pp 65–70
Carmina E et al. The endocrine pattern of late onset adrenal hyperplasia (21-hydroxylase deficiency). J Endocrinol Invest. 1984 Apr;7(2):89-92.
This article was written by Dr. Jeff Donovan, a Canadian and US board certified dermatologist specializing exclusively in hair loss.